Cushing’s Disease 101: Not Rare But Perhaps Rarely Diagnosed

Cushing’s disease, a complex endocrine disorder, is characterized by excessive cortisol production. Often misunderstood and underdiagnosed, Cushing’s disease presents a myriad of symptoms that can significantly impact an individual’s quality of life.
At the Pituitary Disorders Center at Pacific Neuroscience Institute® our expert team provides a holistic approach to treating Cushing’s disease and other pituitary adenomas. In this article we provide a concise overview of the optimal management of Cushing’s disease and highlight both surgical and medical advances.
What is Cushing’s Disease?
Cushing’s disease is caused by a benign (non-cancerous) tumor of the pituitary gland (also known as the master gland) that secretes excess adrenocorticotropic hormone (ACTH). The elevated ACTH levels cause the paired adrenal glands (that sit atop of the kidneys) to produce too much of the stress hormone cortisol. If left untreated, CD and associated elevated cortisol levels can lead to a wide spectrum of problems including obesity, hypertension, diabetes, metabolic syndrome, osteoporosis, impaired immunity, increased risk of infection, sexual dysfunction, depression and impaired memory/concentration. Over time, CD carries a four-fold increased risk of dying, largely related to associated cardiovascular disease, diabetes and obesity.
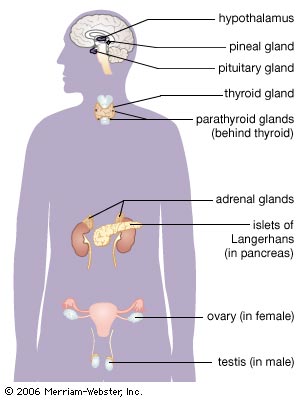
While often described as a “rare” disorder, “relatively uncommon” may be a more apt descriptor of Cushing’s disease. It is much more common in women than men and while it can develop at any age, it tends to manifest between ages 25 to 55 years. The good news is that once Cushing’s disease is diagnosed, endonasal tumor removal performed by experienced pituitary surgeons has a very high success rate and safety record. Overall, 75-80% of patients achieve remission with surgery, and the resultant normalized cortisol levels lead to resolution of many of the associated problems of Cushing disease. If surgery is not initially successful, repeat endonasal surgery, use of medications, focused radiation (radiosurgery) and/or removal of the adrenal glands (bilateral adrenalectomy) can lead to remission in the vast majority of patients. These therapies need to be carefully considered, ideally in a multidisciplinary setting with a team of pituitary specialists.
The two major challenges in Cushing’s disease are:
1. Making the diagnosis and
2. Effectively removing the adenoma and normalizing ACTH and cortisol levels
Herein we describe how these challenges are being addressed.

“I am a critical care nurse by profession, and I knew something was very wrong. Yet after countless labs and multiple MRIs that showed no tumor, it was difficult to find a diagnosis. On a Monday afternoon in September of 2013, I walked into Dr. Daniel Kelly’s office and that day he confirmed I had Cushing’s disease, caused by a pituitary tumor.” – Vanessa B.
Diagnosing Cushing’s Disease
While this may sound relatively simple, clinching a diagnosis of Cushing’s disease can be quite challenging; in fact some endocrinologists consider it one of the toughest diseases to diagnose in all of medicine. Why…?
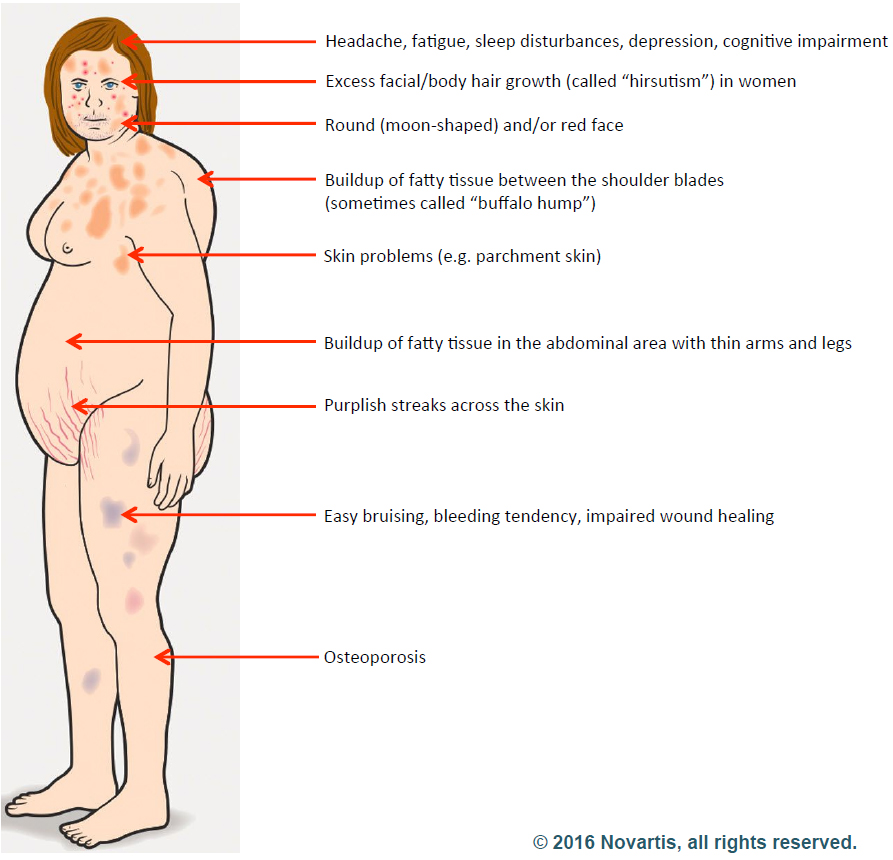
1) No one considered the diagnosis:
Despite a person having developed many of the symptoms, signs and outward appearance of Cushing’s disease, many patients may go undiagnosed for years as their condition worsens. Frequently physicians may not have entertained the diagnosis or simply dismissed the idea given that CD is considered by many to be a “rare disease…” – after all, over 35% of the U.S. adult population is obese (BMI>30) and many physicians simply chalk this up to over-eating and under-activity. We often hear that our patients were told, “Eat less, exercise more, get a personal trainer…” which in many instances they were already doing. In fact, many patients, their family members or close friends are the first to ask “Could this be Cushing’s?” driven to this somewhat obvious question by “research on the internet.”
2) The test results were inconsistent or inconclusive:
Normally, cortisol and ACTH levels fluctuate throughout the day, tending to be higher in the morning and lower in the late afternoon, evening and night. In patients with Cushing’s disease, this diurnal variation is typically lost and the overall quantity of cortisol produced by the adrenal glands is excessive. So endocrinologists over the years have come up with many ingenious ways to accurately measure the excess cortisol. Examples include collecting blood, 24 hour urine and late night salivary samples as well as doing sophisticated sampling of blood near the pituitary gland in the skull base (a procedure called inferior petrosal sinus sampling). In some patients with equivocal results, additional or repeat testing may be required before recommending surgery. It is ideal that an endocrinologist experienced in managing people with Cushing’s disease be in charge of the evaluation process for determining if a patient does or does not have CD.
Surgery and Treatment for Cushing’s Disease
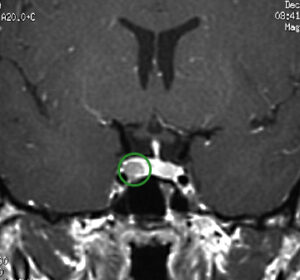
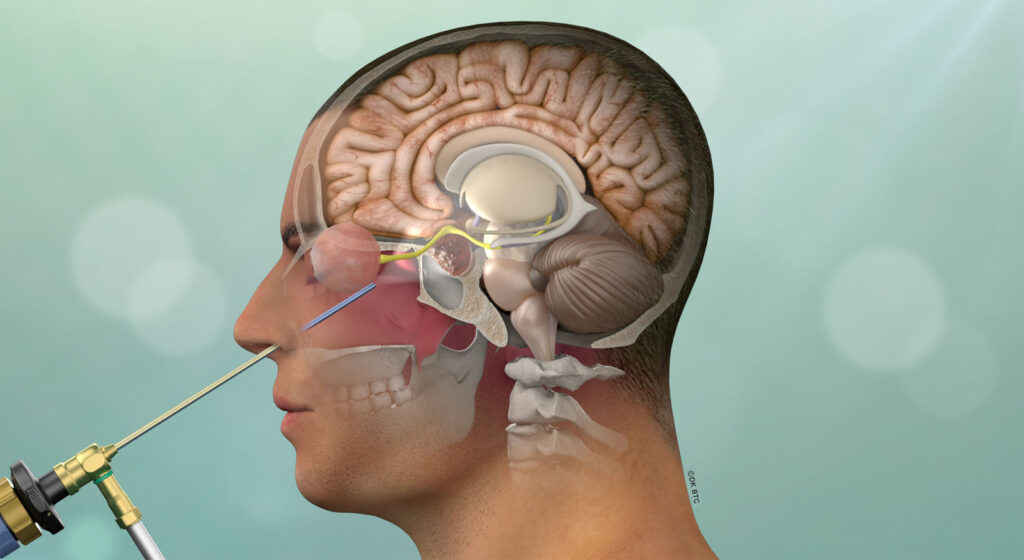
After diagnostic confirmation of Cushing’s disease has been made, endonasal removal of the adenoma and preservation of the normal pituitary gland is the treatment of choice. When performed by an experienced pituitary surgery team, long-term remission rates average close to 80% and the risk of major surgical complications is very low, including a risk of new pituitary gland failure of less than 5%. The success rates are highest (typically over 90%) in patients with a clearly defined non-invasive microadenoma (less than 1 cm in diameter) as seen on MRI. For patients with invasive adenomas or those in which the adenoma is not visible on high-quality pituitary MRI with dynamic sequencing, remission rates are substantially lower (typically ranging from 40-70%).
We routinely monitor blood ACTH and cortisol levels every 8 hours in the immediate post-operative period after surgery. If successful, most patients will have a rapid drop in their ACTH and cortisol levels within 24-48 with the blood cortisol levels dropping to less than 3 – 4 µg/dL, indicating early remission. Most patients with such low cortisol levels feel the “crash” in their stress hormone level with severe fatigue, nausea and joint pains. Once documented, the low cortisol level is treated with an intravenous dose of hydrocortisone and then twice daily oral steroid treatments. Two commonly used steroid replacements are hydrocortisone and prednisone. The typical physiological replacement dose is hydrocortisone 20 mg in the morning and 10 mg in the late afternoon or prednisone 5 mg in the morning and 2.5 mg in the late afternoon. In the first 1-2 months after successful surgery, some patients require slightly higher doses to offset the symptoms of cortisol withdrawal. Patients with successful Cushing’s disease treatment will typically require steroid replacement therapy for at least 6-12 months as the normal physiological feedback loop between the pituitary gland, adrenal glands and hypothalamus gets re-established and the normal ACTH-producing cells of the pituitary gland start producing normal amounts of ACTH.

If a patient is able to be weaned off replacement therapy sooner, that is concern for recurrent Cushing’s disease, which can be monitored by measuring blood and/or salivary cortisol levels and by a 24-hour urinary free cortisol value. Long-term neurosurgical and endocrinological follow-up of CD patients who achieve remission is strongly recommended given that late recurrences can occur, typically within 2-5 years after surgery but as late as 10 years after surgery. We typically follow our patients with blood levels of ACTH and cortisol and a 24-hour urinary free cortisol every 6 months for at least 2-3 years post-surgery and then annually for at least 10 years.
I was thrilled to finally have proof I had Cushing’s, but terrified because I knew I’d have to have brain surgery to remove it. I knew I wanted transsphenoidal surgery – the safest, most successful procedure with the least complications if done by an experienced surgeon. – Elisabeth N.
What if Pituitary Surgery Fails and Cushing’s Disease Persists?
For patients who do not achieve remission of their CD with transsphenoidal surgery, several additional treatment options are available that are often quite effective. These include repeat pituitary surgery, medical therapies with agents such as ketoconazole, pasireotide (Signifor) and mifepristone (Korlym), as well radiosurgery and bilateral adrenalectomy. Such treatments and the order in which they are applied need to be individualized for each patient. In general, however, if pituitary surgery is no longer a viable option, medication therapy is typically considered second-line therapy, followed by radiosurgery or stereotactic radiotherapy; bilateral adrenalectomy is usually considered a last resort, but is highly effective. For more detailed information on these treatment options, there are clinical practice guidelines: Treatment of Cushing’s Syndrome: An Endocrine Society Clinical Practice Guideline published in Lancet Diabetes Endocrinol. 2021 Dec; 9(12): 847–875 by Fleseriu et al.
Being your own advocate to get the diagnosis and treatment right
Given the complexities of diagnosis and treatment of Cushing’s disease, it is critical that patients persist in getting to experienced clinicians in the management of this disorder. We too often see patients who have spent years in search of a diagnosis and getting perhaps well-intentioned but misguided advice and care. Be proactive and seek out a Pituitary Center of Excellence and use available patient and education resources such as these informational sites:
- Pituitary Network Association
- pituitarysociety.org/patient-education
- cushings-help.com
- hormones411.org
Cushing’s Disease Specialists









Pacific Pituitary Disorders Center
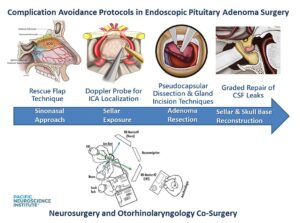
The Pacific Pituitary Disorders Center has a long experience and academic track record in treating patients with pituitary adenomas and Cushing’s disease (CD) in particular. We also treat many patients with recurrent Cushing’s disease. For the last 14 years we have used an exclusively endoscopic endonasal approach given the optimal visualization of the pituitary gland and related skull base anatomy afforded by the hi-definition endoscope. For every case we utilize high-definition cameras and monitors, surgical navigation and Doppler probe for real-time carotid artery localization to maximize effectiveness and safety of the surgery. Our typical pituitary adenoma surgery takes about 3-4 hours and most patients are discharged home a day after surgery.
Schedule a Consultation:
Santa Monica 310-582-7450 | South Bay 424-212-5361
About the Author