
Pituitary Gland Disorders in Women
The pituitary gland may be tiny, but it plays a significant role in a woman’s health. When functioning properly, this master gland regulates essential hormones that influence many of bodily functions, from menstrual cycles to metabolism. Therefore, when pituitary gland disorders arise, they can disrupt reproductive health, energy levels, and overall quality of life. In this article, we’ll explore the role of the pituitary gland in women’s health, common disorders, symptoms to watch for, and how these conditions can be diagnosed and treated.
The Role of the Pituitary Gland in Women’s Health
What is the Pituitary Gland?
The pituitary gland is a pea-sized organ located at a bony structure called the Sella Turcia at the base of the brain, right behind the bridge of the nose. Often referred to as the “master gland,” it controls the production and release of hormones that regulate many crucial body functions. Despite its small size, its impactespecially on a woman’s reproductive system, is powerful.
The Pituitary Gland’s Impact on Female Reproductive Health
In women, the pituitary gland is responsible for producing hormones that are critical for reproductive health, including:
- Follicle-stimulating hormone (FSH) and luteinizing hormone (LH): Regulate the menstrual cycle, ovulation, and fertility.
- Prolactin: promotes lactation after childbirth.
- Adrenocorticotropic hormone (ACTH): Regulates cortisol and other adrenal hormones, such as androgens, from the adrenal glands
- Thyroid-stimulating hormone (TSH): Regulates thyroid functions
- Growth hormone (GH): Supports metabolism, bone density, and muscle strength.
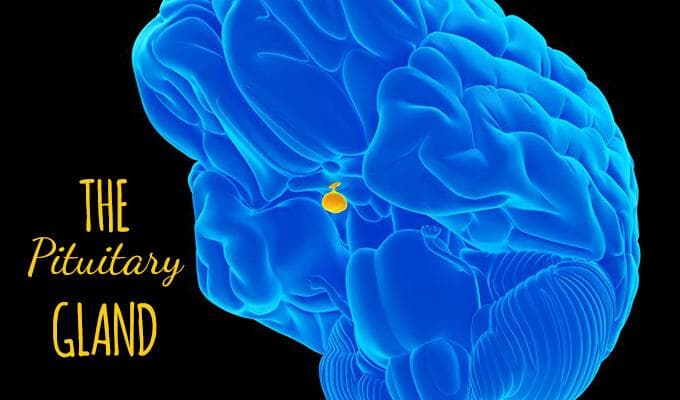
How the Pituitary Gland Regulates Menstrual Cycles, Ovulation, and Pregnancy
A well-functioning pituitary gland ensures regular menstrual cycles and successful ovulation by maintaining a delicate balance of FSH and LH levels. It also helps the body prepare for pregnancy and supports hormonal balance during gestation. Disruption of pituitary function can cause irregular or absent periods, infertility, and pregnancy complications.
What Are Pituitary Disorders and How Do They Affect Women?
Common Pituitary Gland Disorders in Women
Pituitary gland disorders occur when the gland produces too much or too little of certain hormones, often due to benign tumors or other underlying conditions. In women, these disorders can lead to reproductive challenges, metabolic changes, and emotional well-being issues. Learn about a hormonal evaluation at Pacific Neuroscience Institute®.
Examples of hypersecretion
- Prolactinoma: A noncancerous tumor that causes excess prolactin production, leading to LH and FSH suppression, which causes irregular periods, infertility, and unexpected breast milk production (galactorrhea).
- Cushing’s Disease: Caused by a tumor leading to excessive ACTH production and elevated cortisol levels. Women may experience weight gain, menstrual irregularities, and mood swings.
- Acromegaly: A result of excess growth hormone, leading to enlargement of the hands, feet, and facial features. It can also disrupt menstrual cycles and increase diabetes risk.
What is Hypopituitarism, and How Does it Impact Women?
Pituitary Disease and Hypopituitarism
Hypopituitarism occurs when the pituitary gland fails to produce sufficient amounts of one or more hormones. It can happen due to pituitary adenomas or other sellar masses that cause mass effect, either leading to dysfunction of the normal cells, or by increasing prolactin levels which suppress LH and FSH hormones In women, this can disrupt menstrual cycles, lower estrogen and progesterone levels, impair fertility, and impact overall health.

Impact on Estrogen, Progesterone, and Prolactin
- Low estrogen: Hot flashes, vaginal dryness, and mood swings
- Low progesterone: Irregular menstrual cycles and difficulty maintaining pregnancy
Low prolactin: Challenges with breast milk production postpartum
Common Hypopituitarism Symptoms in Women
- Irregular or absent periods (amenorrhea)
- Difficulty conceiving
- Low libido
- Fatigue and low energy
- Weight changes
- Change in hair patterns
- Lactation difficulties after childbirth
The Long-Term Effects of Untreated Pituitary Disease
Without appropriate diagnosis and treatment, pituitary gland disorders can cause permanent infertility, osteoporosis (due to low estrogen levels, low GH, or high cortisol), metabolic conditions, cardiovascular complications, and a significant decrease in quality of life.
Symptoms of Pituitary Gland Problems in Women
Recognizing symptoms early is key to receiving timely treatment. Common signs of pituitary gland issues in women include:
- Irregular or missed periods
- Fatigue
- Unexpected weight gain or loss
- Headaches or vision problems (due to tumor pressure)
- Mood changes, such as irritability, depression, or anxiety
- Acne
- Hirsutism (excessive hair growth)
- Breast milk production without pregnancy (galactorrhea)
Diagnosing and Treating Pituitary Gland Disorders
To diagnose pituitary gland issues in women, physicians often obtain:
- Thorough history taking and physical exam
- Hormonal Blood Tests: To measure levels of FSH, LH, estradiol, prolactin, thyroid functions, growth hormone, IGF-1, ACTH, and cortisol.
- Imaging Techniques: dedicated pituitary MRI to identify pituitary tumors that could affect reproductive health and hormone production.
Treatment Options for Pituitary Disorders
Medical Treatments
Many pituitary disorders can be successfully managed with medications:
- Restoring Hormonal Deficiencies: Using estrogen, progesterone, thyroid hormones, growth hormone, or corticosteroids as needed.
- Dopamine Agonists: To shrink prolactinomas and lower prolactin levels.
- Specific treatments to control hypersecretion of ACTH or GH
Surgical Interventions for Pituitary Gland Tumors
When tumors affect surrounding structures, is functionally active or disrupt hormone production, surgery may be necessary. Most procedures are minimally invasive endonasal endoscopic approaches and involve removing the tumor through the nose, which helps restore normal hormonal balance and can improve reproductive outcomes.
Long-Term Monitoring
After treatment, regular follow-up appointments are essential. Monitoring symptoms and hormone levels helps maintain balance and manage reproductive health, particularly for women considering pregnancy and in other periods in their lives, such as post menopause.
Managing Life with a Pituitary Disorder
Women living with a pituitary disorder can take proactive steps to support their health:
- Lifestyle Changes: Maintain a balanced diet, exercise regularly, and manage stress to promote hormonal health.
- Regular Monitoring: Keep track of menstrual cycles, hormone levels, bone density, and cardiometabolic health.
- Support Networks: Seek out women’s health support groups and counseling to manage emotional and fertility-related challenges.
Pituitary gland disorders can significantly impact a woman’s reproductive health, energy levels, and quality of life. Early recognition, accurate diagnosis, and personalized treatment are essential for managing these conditions. Whether dealing with prolactinomas, Cushing’s disease, or hypopituitarism, expert care can help restore hormonal balance and improve well-being.
If you are experiencing symptoms of a pituitary disorder, reach out to specialists at the Pituitary Disorders Center at Pacific Neuroscience Institute® to schedule an appointment and take an important step toward better health.
About the Author
