
Craniopharyngioma: The Most Formidable of Intracranial Tumors
Treatment Challenges and Advances in 2017
Almost 80 years ago, the father of modern neurosurgery, Dr. Harvey Cushing, declared craniopharyngiomas “the most formidable of intracranial tumors.” Back in 1939, before the use of the surgical microscope, the endoscope, modern anesthetic techniques and hormone replacement therapies, Cushing learned that surgery for these tumors was extremely challenging and risky.
Fast forward to the present, and Cushing’s characterization of these tumors remains accurate, however, our comprehensive team approach for patients with craniopharyngiomas has changed dramatically and overall outcomes are much improved. Herein we describe our present management of these uncommon tumors, including diagnosis, surgical approach, radiation therapy, hormone replacement and exciting new targeted therapies on the horizon.
OVERVIEW
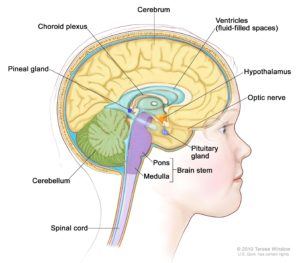
Although classified as benign (non-cancerous), craniopharyngiomas pose a great challenge in management because they typically adhere to the pituitary gland and stalk, optic nerves and optic chiasm, surrounding blood vessels as well as the brain itself. They are thought to arise from developmental remnants of the craniopharyngeal duct or Rathke’s pouch. They can occur at any age but are most common in childhood and adolescence and in adults over age 50. They account for 10-15% of sellar and suprasellar tumors (tumors that occur in and above the pituitary gland) and over 50% of such tumors in children. They are usually not discovered until they impinge upon important structures, and are frequently quite large (over 3 cm) when detected.
SYMPTOMS
The most common symptoms of craniopharyngiomas are related to loss of hormonal function (hypopituitarism), including low energy, low libido, loss of menstrual periods in women and weight gain, growth failure in children), loss of vision, and headache. Some craniopharyngiomas may also become quite large, blocking flow of cerebrospinal fluid (CSF), leading to hydrocephalus with changes in mental state and cognition.
DIAGNOSIS
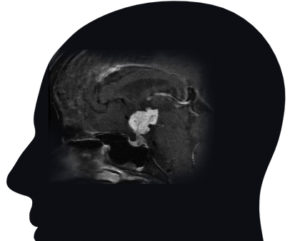
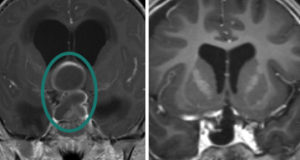
Craniopharyngiomas are typically diagnosed on MRI or CT scan, and are usually distinguishable from the more common pituitary adenomas, Rathke’s cleft cysts and meningiomas that might occur in the region around the pituitary gland and skull base. Craniopharyngiomas typically have both solid and cystic (fluid-filled) components, and many have calcification as seen on CT scan. Complete pituitary hormonal testing is also essential given that a majority of patients will have hormonal deficits. Similarly, most patients will need formal visual field and acuity testing to assess the impact of the tumor on vision and eye movements.
TREATMENT
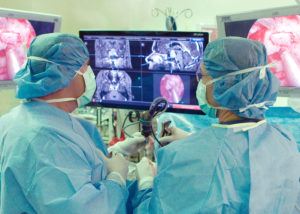
First-line treatment for craniopharyngiomas is maximal safe surgical removal, although in a minority of mostly cystic craniopharyngiomas, simple cyst drainage may be the first treatment. Second-line treatment is typically focused radiotherapy or radiosurgery. Given that most craniopharyngiomas arise along the pituitary stalk, the most common surgical approach is the endoscopic endonasal route via the nostrils. If pituitary gland function is intact, every attempt is made to preserve the integrity of the pituitary gland and stalk and its connection to the hypothalamus. Alternatively, for tumors that extend far off the midline and for some recurrent craniopharyngiomas, a supraorbital “eyebrow” craniotomy can be used. Several examples of these approaches for craniopharyngioma are shown in our video library.
Given the tendency of craniopharyngiomas to adhere to critical surrounding blood vessels, cranial nerves and the brain itself, particularly the hypothalamic region, “near complete” or subtotal removal occurs in a majority of patients while complete (100%) removal occurs in less than half of cases. This approach also has a higher likelihood of preserving pituitary gland function. Typically, if small bits of tumor are left behind, these are closely monitored, and then treated with focused stereotactic radiotherapy or radiosurgery, if they grow on subsequent MRI. This treatment formula of maximal safe surgery followed by focused radiation if the residual tumor grows, has a high success rate in our hands as well as at other high-volume Pituitary Centers of Excellence. A relatively new discovery is that a subset of craniopharyngiomas described as “papillary”, typically have a BRAF mutation that can be a potential target for a BRAF inhibitor drug. This new BRAF “targeted therapy” for recurrent papillary craniopharyngiomas is the focus of a new clinical trial for patients with tumors not controlled with surgery and radiation. Hopefully other new molecular therapies are also on the horizon for these tumors.
FOLLOW-UP
Craniopharyngiomas patients need long-term neurosurgical and endocrinological follow-up with regular MRIs and hormonal evaluations, as late recurrences can occur. We typically follow our patients with regular MRIs every 3-6 months for at least 2-3 years post-surgery and then annually for at least 10 years.
OUR EXPERIENCE IN CRANIOPHARYNGIOMA
With an overall series of over 1800 endonasal operations for all types of pituitary and related skull base tumors, at Pacific Pituitary Disorders Center, we have a long experience and academic track record in treating these highly complex tumors using a team approach of neurosurgery, ENT, endocrinology, radiation oncology, neuro-ophthalmology and neuro-oncology. For the last 9 years, we have used an exclusively endoscopic endonasal approach for all pituitary tumors including craniopharyngiomas, given the optimal visualization of the pituitary gland and related skull base anatomy afforded by the hi-definition endoscope. We also use surgical navigation and the Doppler probe for real-time carotid artery localization to maximize effectiveness and safety of the surgery. In our series of 70 craniopharyngioma surgeries, of the more recent fully endoscopic surgeries, 80% of patients undergoing first-time surgery have achieved a near total or gross total tumor removal with a very low complication rate and a high rate of gland and stalk preservation. Our typical endonasal craniopharyngioma surgery takes about 5-6 hours and most patients are discharged home on day 2 after surgery. To learn more about how endoscopic pituitary surgery for craniopharyngiomas and other pituitary tumors is performed and our expert team, visit us at www.pacificpituitary.org. Also see our pituitary-related publications.
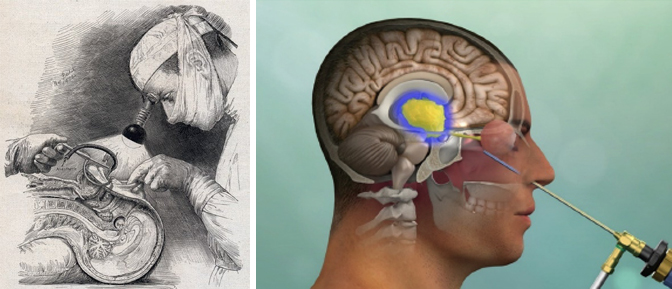
Banner image: Dr. Harvey Cushing performing sublabial transsphenoidal surgery with a head lamp, drawn in 1912 by Max Brodel, contrasted with current endonasal transsphenoidal approach for craniopharyngioma illuminated with high definition endoscope.

Daniel F. Kelly, MD, is the Director of the Pacific Neuroscience Institute in Santa Monica, CA. Considered to be one of the top neurosurgeons in the US, Dr. Kelly is internationally recognized in the field of minimally invasive keyhole surgery for brain, pituitary and skull base tumors. He continues to focus his efforts on advancing innovative treatments for patients, providing fellowship training in minimally invasive neurosurgery, and patient education and support.
About the Author

Last updated: September 8th, 2020