Overview
What is a Craniopharyngioma?
Overview
A craniopharyngioma is a rare type of benign brain tumor that develops near the pituitary gland at the base of the brain. This tumor originates from remnants of embryonic tissue that are normally present during early development.
Craniopharyngiomas are usually not discovered until they impinge upon important structures around them, and are frequently quite large (over 3 cm) when detected. Despite being non-cancerous, craniopharyngiomas can cause significant health problems due to their location and potential impact on nearby structures. These tumors tend to become adherent to structures around the pituitary gland and stalk, including the optic nerves, optic chiasm, intracranial arteries and the brain itself.
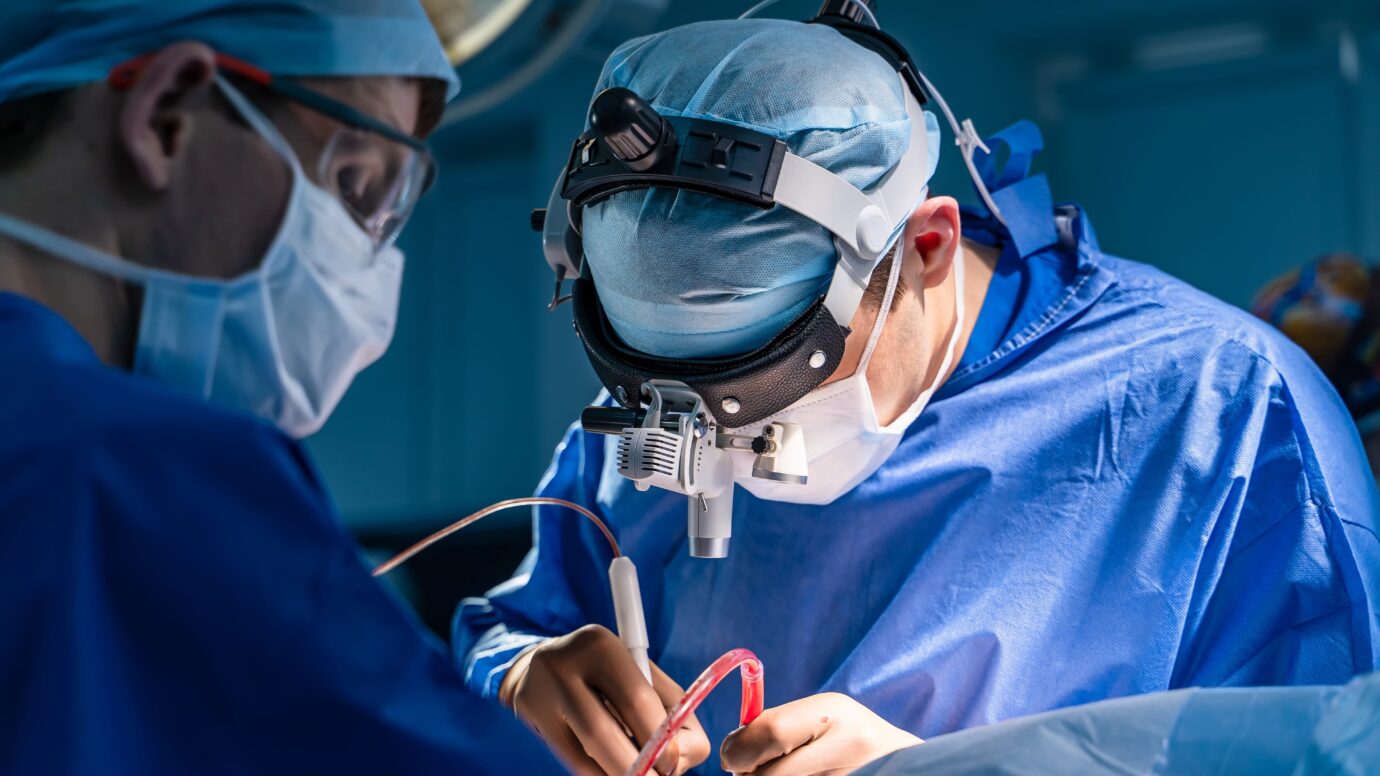
Surgical treatment: The initial optimal treatment for craniopharyngiomas is maximal safe surgical removal. Fortunately for most patients, craniopharyngiomas can be removed through a keyhole route via the nose using an endoscopic endonasal approach or an eyebrow craniotomy.
Hormonal replacement: Many if not most patients with a craniopharyngioma will need some form of pituitary hormone replacement.
Radiation therapy: A significant minority may need radiosurgery or focused radiotherapy to prevent tumor regrowth.
Other targeted therapies: Given that some craniopharyngiomas cannot be removed completely by surgery or controlled with radiation therapy, tumor genomic profiling is increasingly used to find alternative targeted therapies for these challenging tumors. In the case of papillary craniopharyngiomas, we also profile these tumors looking specifically for the BRAF mutation that may allow treatment with a targeted drug called a BRAF inhibitor.
Who is Affected?
Craniopharyngiomas occur most commonly in childhood and adolescence and in later adult life, after age 50. They account for 10-15% of sellar and suprasellar tumors (tumors that occur in and above the pituitary gland) and 50-60% of sellar and suprasellar tumors in children.