
Chordoma: A Rare Bone Cancer of the Skull Base and Spine
Chordoma is a rare, low-grade cancer that can occur anywhere along the spine or skull base. These tumors are believed to arise from the notochord, a vestigial structure involved in early spinal column development.
Chordomas are extremely rare, affecting approximately 1 in 1,000,000 people per year, and account for less than 4% of all primary bone cancers. They most commonly present between 40 and 70 years of age and occur more frequently in men than women.
Although chordomas rarely metastasize, they are locally invasive and aggressive, often affecting nearby critical neurological structures.
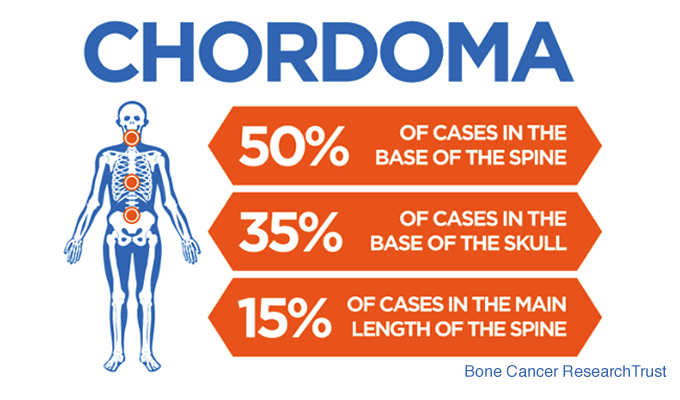
Where Do Chordomas Occur?
Chordomas most commonly arise in three regions:
- The skull base (approximately one-third of cases)
- The mobile spine
- The sacrum
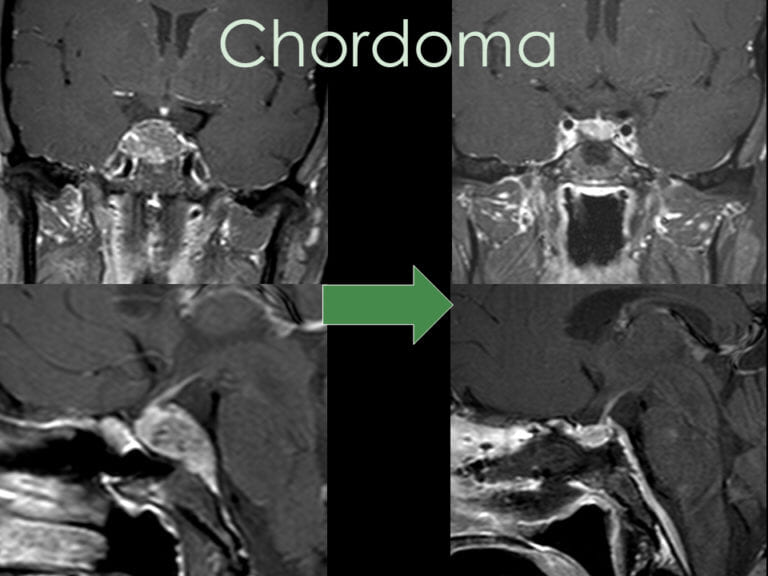
Skull Base (Clival) Chordomas
Many skull base chordomas originate in the clivus, a bony junction between the sphenoid and occipital bones. As these tumors grow, they can destroy bone and extend into the brain, cranial nerves, and soft tissues.
Chordomas may breach the dura mater (outer brain covering), placing pressure on the brainstem and cranial nerves, or extend into the nasal sinuses, leading to obstruction, sinus infections, bleeding, or Eustachian tube dysfunction.
Symptoms of Chordoma
Symptoms of chordoma vary depending on tumor size and location, and often develop gradually.
Common Symptoms of Skull Base Chordoma
- Persistent headaches (often resembling sinus headaches)
- Double vision (diplopia)
- Abnormal eye movements
- Hearing difficulties
- Nosebleeds (epistaxis)
- Chronic sinus infections
Cranial Nerve and Brainstem Chordoma Symptoms
The most commonly affected cranial nerve is the abducens nerve, which controls eye movement. Other nerves that may be involved include:
- Trigeminal nerve (facial sensation)
- Facial nerve (facial movement)
- Vestibulocochlear nerve (hearing and balance)
- Optic nerves (vision loss)
- Hypoglossal nerve (tongue movement)
Severe brainstem compression may result in:
- Difficulty swallowing (dysphagia)
- Hoarse voice (vocal cord dysfunction)
- Weakness in arms or legs
- Balance and coordination problems
Pituitary Dysfunction
Because the pituitary gland lies at the top of the clivus, skull base chordomas may compress or displace it. This can lead to hormonal dysfunction, including:
- Hypothyroidism
- Hypocortisolism
- Fatigue
- Weight gain
- Loss of menstrual cycle
- Impotence or infertility
Some chordomas are discovered incidentally on imaging performed for unrelated reasons. Even asymptomatic lesions require careful evaluation due to their potential for growth.
How is Chordoma Diagnosed?
Chordomas are initially identified through imaging studies, most commonly:
- CT scans, often ordered for sinonasal symptoms
- MRI with IV gadolinium contrast, using thin cuts through the clivus (often called a pituitary protocol)
Additional Diagnostic Evaluation
If imaging shows compression of the pituitary gland or optic chiasm, further evaluation may include:
- Endocrinology assessment
- Ophthalmology evaluation
- Pituitary hormone testing
- Visual field testing and optical coherence tomography (OCT)
Biopsy and Pathology
Because many tumor types can arise in the skull base, a biopsy is almost always required to establish a definitive diagnosis. This is typically performed by an ENT surgeon during an outpatient procedure. Pathologic confirmation of chordoma is essential to guide treatment.
Treatment of Chordoma at Pacific Neuroscience Institute
Surgical Treatment
The gold standard for chordoma treatment is surgical resection of the tumor. Most skull base chordomas are midline tumors and are best approached through a minimally invasive approach such as an endoscopic endonasal transclival approach through the nostrils.
Tumors with lateral extension may require alternative or combined approaches, such as a retrosigmoid craniotomy from behind the ear. The goal is maximal safe resection, prioritizing preservation of neurological function over aggressive removal.
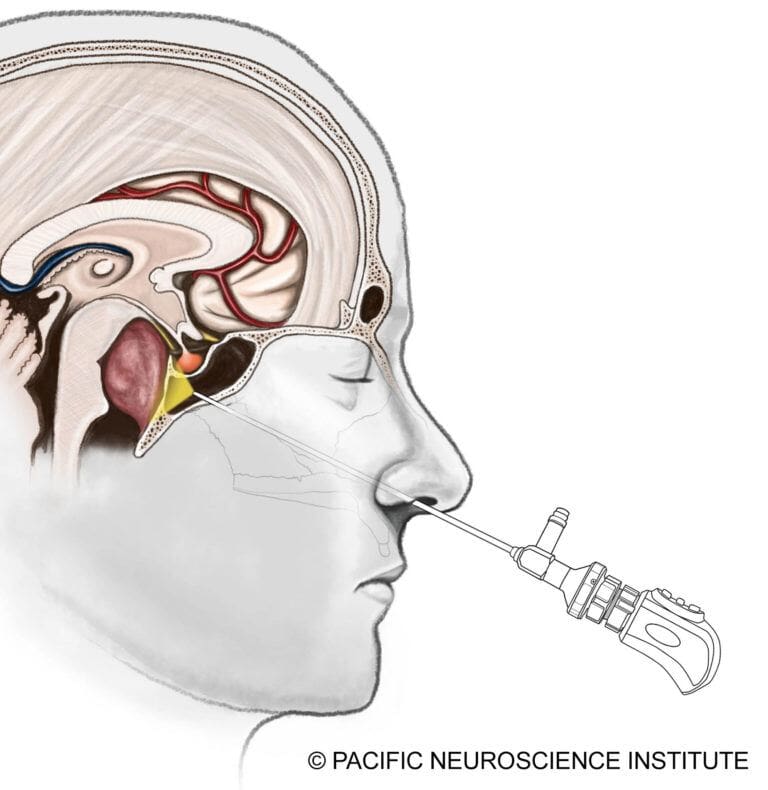
Advances in Endoscopic Surgery
Modern endonasal surgery has become significantly safer through:
- Surgical navigation (“GPS for surgery”)
- Doppler ultrasound monitoring
- Continuous cranial nerve monitoring
The development of the nasoseptal vascularized flap has dramatically reduced postoperative cerebrospinal fluid (CSF) leaks, which can otherwise lead to meningitis.
Radiation Therapy
If residual tumor remains or regrowth occurs, additional treatment may include:
- Stereotactic radiosurgery
- Proton beam radiation
- Or a combination of both
These therapies provide approximately 80% tumor control, with proton therapy offering advantages in select cases depending on tumor location.
Medical and Targeted Therapies
While no medication has proven curative for chordoma, genetic sequencing may identify actionable targets. At Saint John’s Cancer Institute, ongoing studies are evaluating novel therapies for chordoma patients, and eligible patients may participate in clinical trials.
Comprehensive Care for Chordoma Patients
Chordomas are rare, slow-growing but aggressive tumors that often present with subtle symptoms. Management typically involves biopsy, minimally invasive surgical resection, and, when necessary, radiation therapy. Emerging research into targeted therapies offers hope for patients with recurrent disease.
At Pacific Neuroscience Institute patients benefit from a multidisciplinary approach involving neurosurgeons, ENT surgeons, endocrinologists, ophthalmologists, radiation oncologists, and neuro-oncologists.
For more information or to request a consultation with one of our experts, contact us at 310-582-7450.
Chordomas can present with subtle symptoms and have an insidious growth rate. Treatment options are tumor biopsy followed by minimally invasive tumor resection. Some patients may require radiation therapy if there is tumor growth following surgery. Leading-edge research has identified several potential medications that may offer some benefit for patients with recurrent chordomas. We at PNI and the Saint John’s Cancer Institute are able to offer a comprehensive approach to chordoma patients with our team of neurosurgeons, ENT surgeons, ophthalmologists, endocrinologists, radiation oncologists and neuro-oncologists.
For more information or for a consultation with one of our experts, contact us at 310-582-7450.
About the Author
