
Acromegaly
Endoscopic endonasal surgery is a preferred first-line treatment for patients with Acromegaly
On this page: Overview | Symptoms | Diagnosis | Treatment | Doctors and Specialists
Acromegaly is caused by a growth hormone (GH) secreting pituitary adenoma.
For the great majority of patients with acromegaly, transsphenoidal tumor removal (via the nose) is the initial treatment of choice. Fortunately, there have been major technical advances in this surgical approach over the last two decades, including use of high-definition endoscopic visualization, surgical navigation techniques and more effective skull base closure methods. As a result, in experienced hands, this procedure has a relatively high success rate and low complication rate. For patients who are not cured by surgery alone, medical therapy with agents such as octreotide and lanreotide are considered 2nd-line therapy; stereotactic radiosurgery (SRS) or stereotactic radiotherapy (SRT) are considered reasonable 3rd-line therapies.
Our Approach to Hormone Care
At Pacific Pituitary Disorders Program, we have one of the world’s largest experiences in endoscopic endonasal transsphenoidal surgery. By incorporating cutting-edge technology and instrumentation with proven surgical experience of over 2,000 endonasal surgeries, our acromegaly specialists, make surgery safer, less invasive and more effective. Our doctors and surgeons practice at award winning hospitals throughout Los Angeles.
Center Director and Neurosurgeon Dr. Daniel Kelly and his neurosurgical partner Dr. Garni Barkhoudarian and ENT Surgeons Drs. Chester Griffiths and Kian Karimi, have a large surgical experience treating patients with newly diagnosed acromegaly as well as patients with persistent or recurrent acromegaly after prior surgery.
For patients with invasive GH-secreting tumors that cannot be completely removed by surgery, we have highly experienced endocrinologists as well as clinical trials for patients with persistent acromegaly.
- See our pituitary adenoma publications
- Review the latest Clinical Practice Guidelines for Acromegaly
What is Acromegaly?
Acromegaly is caused by a growth hormone (GH) secreting pituitary adenoma in over 99% of cases.
These benign and slow growing tumors are not rare. Acromegaly patients comprise a large portion of any busy pituitary center of excellence like ours here at PNI. Unfortunately, many patients go undiagnosed for many years. However, once diagnosed, treatment with endoscopic endonasal surgery is generally highly effective in experienced surgeon’s hands, although a minority of acromegalic patients may also need medical therapies to lower GH and IGF-1 levels. This reality is why such close cooperation is essential between neurosurgery and endocrinology in managing patients with acromegaly.
The problems associated with acromegaly include the effects of abnormally high GH and insulin-like growth factor-1 (IGF-1) levels and in some instances by the tumor compressing the normal pituitary gland and optic nerves.
Untreated acromegaly is a serious condition that can cause dramatic bone and soft tissue changes and serious cardiovascular problems. If the tumor develops before bone growth is completed in adolescence, gigantism results.
Because of the serious changes resulting from GH excess, effective treatment is essential.
Acromegaly Causes
The cause of acromegaly, like for many tumors, is not completely understood. Most pituitary adenomas secreting excess GH and causing acromegaly are caused by mutations in the pituitary cells called somatotrophs.
Acromegaly Symptoms
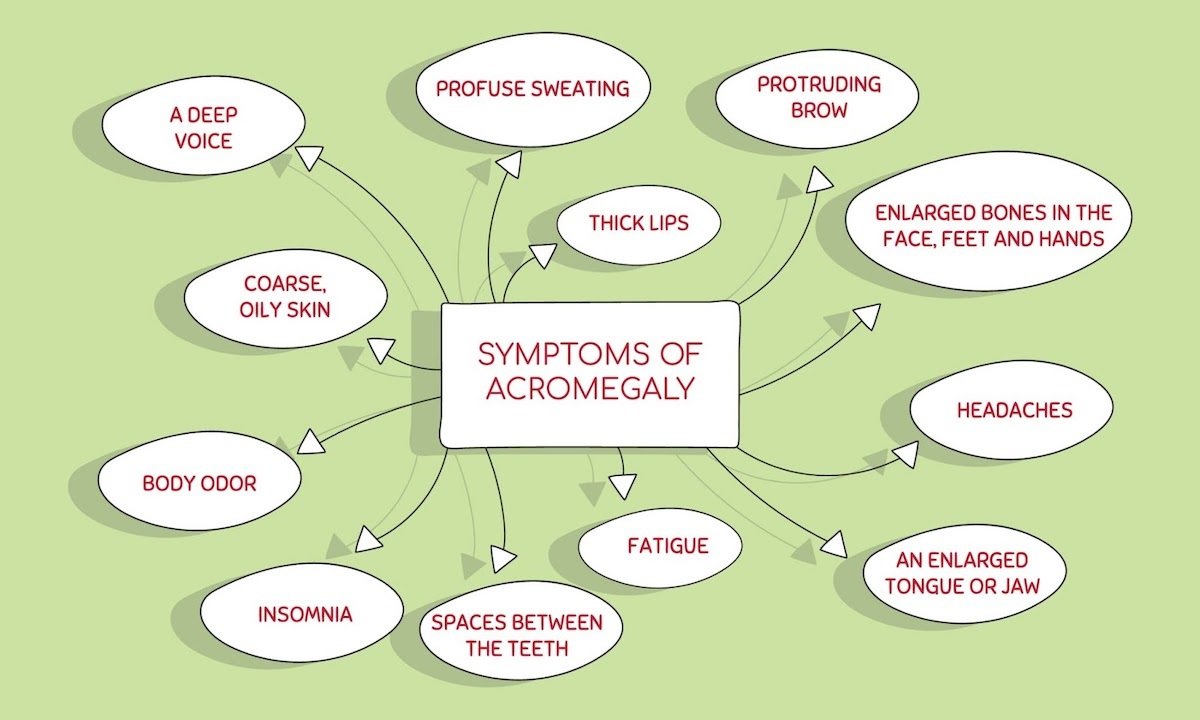
The most obvious symptoms of acromegaly and excess GH are external physical changes that often include:
- Enlargement of the hands (increase in ring size) and feet (increased shoe size)
- Frontal bossing (enlargement of the forehead) and prognathism (jaw enlargement)
- Development of an underbite, spreading teeth, an enlarging tongue
- Increased snoring and sleep apnea
- Carpel tunnel syndrome
- Excessive sweating
Additional serious problems may include:
- Hypertension
- Diabetes mellitus
- An increased risk of colon cancer
With GH-secreting macroadenomas, there may be other symptoms like:
- Visual loss
- Headaches
- Pituitary gland failure
- Fatigue
- Depression
- Impotence
- Loss of libido in men
- Menstrual irregularities
- Galactorrhea (milk discharge from the breast) in women

Acromegaly Diagnosis
Comparing old and recent photographs will often demonstrate gradual but dramatic changes in facial appearance.
However, acromegaly is diagnosed by:
- Growth Hormone Evaluation: Documenting elevated levels of both GH and IGF-1. An oral glucose tolerance test is often used to confirm excess GH production.
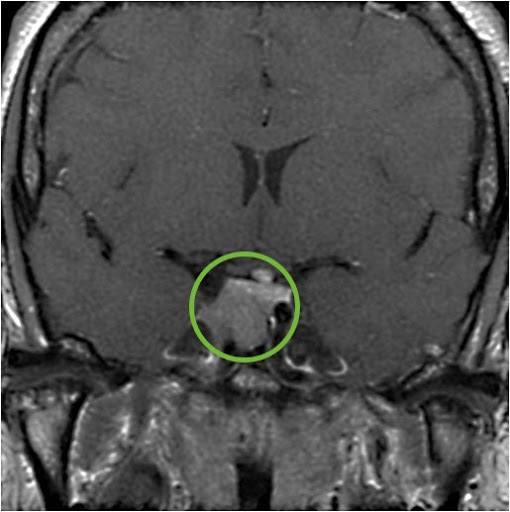
- MRI Imaging: Following hormonal testing that confirms acromegaly, an MRI of the pituitary should be performed to confirm the presence of a pituitary adenoma.
For patients with visual complaints, formal visual field testing by an ophthalmologist or neuro-ophthalmologist should be performed.
Recently Diagnosed?
If you have been recently diagnosed with acromegaly and would like to get more information or a second opinion please contact the Pacific Pituitary Disorders Center at 310-582-7450
Acromegaly Treatment
Acromegaly Surgery
Surgical removal is considered first-line treatment for GH-secreting acromegaly.
Endoscopic Endonasal Surgery for Treatment of Acromegaly
Because of improved tumor visualization, the endoscopic endonasal approach is rapidly becoming the preferred method for removal of most pituitary adenomas, including GH-secreting adenomas.
Long-term remission of acromegaly is often not possible in patients with large or invasive macroadenomas. However, in such invasive tumors, removal of the great majority of the tumor can greatly improve problems associated with acromegaly (visual loss, pituitary gland dysfunction and headache) and typically improves hypertension, diabetes and soft tissue swelling.
Additionally, such maximal tumor debulking improves the chances of achieving remission with medical therapy using medications such as lanreotide or octreotide. After surgery, it typically takes at least 4-6 weeks for the IGF-1 level to reach its lowest level due to its very long metabolic half-life.
Learn more about endoscopic endonasal surgery
What medication is used to treat acromegaly?
For patients with persistent GH and IGF-1 elevations 3 months of more after surgery, octreotide or lanreotide treatments are generally indicated.
Octreotide (given three times a day by injection or by one monthly injection), or lanreotide (deep subcutaneous injections every 4 weeks) achieve long-term suppression of GH in about 70% of patients.
Lanreotide and octreotide also cause tumor shrinkage in 30-50% of patients, and improve soft tissue swelling, headache, joint pains and sleep apnea.
Preoperative use of octreotide and lanreotide may facilitate tumor removal and lessen risks of general anesthesia.
Side effects may include loose stools, malabsorption, cholelithiasis (gall stones), and local pain at the injection site. Pegvisomant, a GH receptor antagonist, is also effective in lowering IGF-1 levels although it does cause an elevation in GH levels.
In patients who have GH-secreting adenomas that also co-secrete prolactin, the dopamine agonists cabergoline or bromocriptine can be combined to help normalize GH and prolactin levels and halt tumor growth.
Stereotactic Radiosurgery (SRS) or Stereotactic Radiotherapy (SRT) for Acromegaly
For patients with uncontrolled acromegaly after surgery and medical therapy, SRS (one dose) or SRT (multiple doses), can be used to deliver precise radiation directly to the tumor.
These techniques are effective in lowering GH and IGF-1 levels and stopping tumor growth in approximately 40-60% of patients. However, the lowering of GH and IGF-1 levels takes longer with SRT (average 7 years) compared to SRS (average 18 months). Pituitary gland failure can often occur in the years after SRS or SRT. Complications such as visual loss are rare with either SRS or SRT.
Patient Outcomes
Long-term remission is seen in 80-90% of patients with microadenomas and in 40-60% of patients with macroadenomas or invasive adenomas. In general, the higher the pre-operative GH level and the larger the tumor, the lower the chance for cure or long-term remission.








